Biomolecular 'Google Maps'
CLEM / multi-scale, multi-modal microscopy faster imaging / digital pathology /
brain imaging / multi-beam SEM |
(Ultra)fast electron microscopy
Time-resolved cathodoluminescence
beam blankers / laser pump electron probe carrier dynamics / nanophotonics |
Light-electron-matter interactions
Electron-beam induced superresolution
low-voltage SEM / beam damage electron-molecule dynamics |
Biomolecular 'Google Maps' - multi-scale multi-modal microscopy
Molecular reactions dictate life at the level of enzymes, cells, organs, and organisms, compassing many orders of magnitude in length scales (in humans from nm to m). The heterogeneity between building blocks such as biomolecules and cells ultimately determines physiological functions, such as cell signalling cascades, growth, metabolism. To visualize how molecules are structured within an organism and how the heterogeneity arises during development is an essential step in understanding biological functions and how dysfunctions occur during disease. Molecules can be visualized using fluorescence microscopy (FM), but with FM the structural background remains invisible. EM can image structural heterogeneity in cells and tissue with nanometer-resolution, but it leaves the molecular content dark and throughput is too low to routinely embed local images in the multi-scale context.
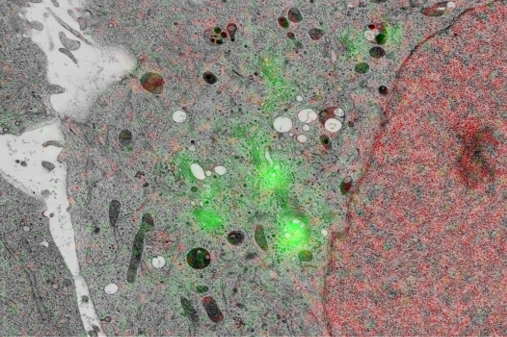
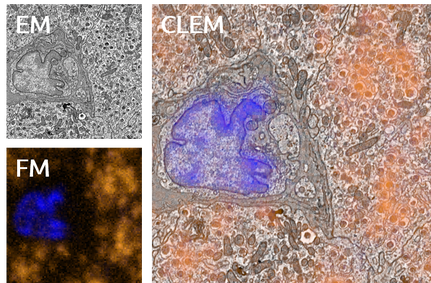
With integrated microscopy, FM images can be routinely embedded in the structural context obtained with EM. We use cathodoluminescence generated in the sample-supporting substrate (ITO-coated glass) to register FM and EM images without the need for fiducial markers, in automated and unbiased fashion with a consistent, high accuracy (<50nm under normal operation, ~5nm with FM-EM distortion correction) throughout the sample. Thus, EM images are directly colored with molecular content, like in the left image. We extent this to large-scale and volume imaging using serial sections in an approach called integrated correlative array tomography (i-CAT).
We develop techniques to enhance throughput in the entire EM pipeline: sectioning, acquisition, and data handling and interpretation. We develop faster detection methods for large-scale imaging. We apply i-CAT for fluorescence-guided EM acquisition, where the microscope itself decides, based on fluorescence expression, which areas to scan at high magnification. Our integrated microscopy platform is used for transmission detection in the Delft multibeam electron microscope that was developed in the Kruit group. We aim to achieve >100x faster acquisition, meaning that a high-resolution partial zebrafish brain acquisition that would otherwise take a full year of uninterrupted imaging, could now be done over a weekend. We apply our technology for digital pathology with EM (with the Giepmans group at UMC Groningen), imaging brain development in zebrafish larvae (with the Carroll group), and 3D tumor reconstruction (with Klumperman and Liv at UMC Utrecht).
With integrated microscopy, FM images can be routinely embedded in the structural context obtained with EM. We use cathodoluminescence generated in the sample-supporting substrate (ITO-coated glass) to register FM and EM images without the need for fiducial markers, in automated and unbiased fashion with a consistent, high accuracy (<50nm under normal operation, ~5nm with FM-EM distortion correction) throughout the sample. Thus, EM images are directly colored with molecular content, like in the left image. We extent this to large-scale and volume imaging using serial sections in an approach called integrated correlative array tomography (i-CAT).
We develop techniques to enhance throughput in the entire EM pipeline: sectioning, acquisition, and data handling and interpretation. We develop faster detection methods for large-scale imaging. We apply i-CAT for fluorescence-guided EM acquisition, where the microscope itself decides, based on fluorescence expression, which areas to scan at high magnification. Our integrated microscopy platform is used for transmission detection in the Delft multibeam electron microscope that was developed in the Kruit group. We aim to achieve >100x faster acquisition, meaning that a high-resolution partial zebrafish brain acquisition that would otherwise take a full year of uninterrupted imaging, could now be done over a weekend. We apply our technology for digital pathology with EM (with the Giepmans group at UMC Groningen), imaging brain development in zebrafish larvae (with the Carroll group), and 3D tumor reconstruction (with Klumperman and Liv at UMC Utrecht).
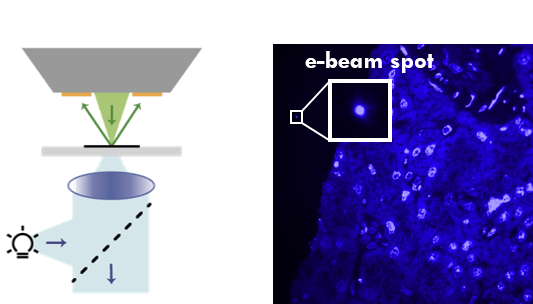
Cathodoluminescence registers electron beam to FM image
|
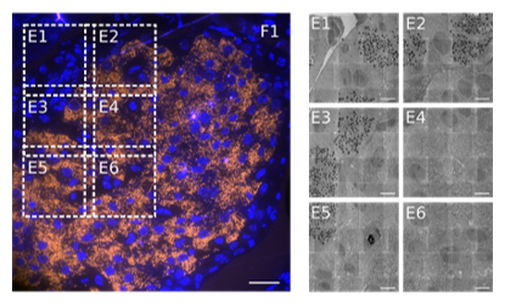
Large-scale integrated correlative array tomography
|
(Ultra)fast microscopy
The key ingredient to adding temporal resolution in an electron microscope is the generation of a pulsed beam of electrons. Contrary to the often used photo-emission sources, we use beam blanking to generate the pulsed beam. Beam blanking has the advantages that (i) it can be easily implemented in an electron microscope (often blankers are a standard asset to the microscope) and does not require modifications to the high-voltage source like in the case of laser-triggered photo-emission, (ii) it preserves the brightness of the electron source, and (iii) it is highly stable over long measurement times (which are often needed as the current is substantially reduced in a pulsed beam compared to continuous-beam operation). With a standard commercial beam blanker, we achieved <90ps pulses and we have developed concepts for laser-triggered blankers that could potentially generate shorter pulses.
|
Electron pulse creation with a beam blanker
|
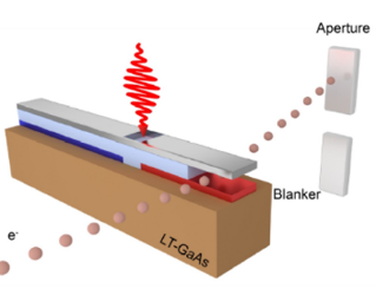
Proposed scheme for a laser-triggered beam blanker
|
We use time-resolved electron microscopy to map the flow of energy in a material after photon excitation. Once a pulsed electron beam has been created, two types of temporal experiments can be implemented. First, the electron beam can be used to excite cathodoluminescence in the sample material. Detection of the emitted photons with a time-correlated detector that further receives a trigger signal from the pulse generator driving the blanker, allows to build a histogram of photon arrival times. This can be used to reveal carrier migration times, or, when an active phosphorescent of fluorescent material is used, to measure lifetimes with spatial resolution far below the diffraction limit. These provide a direct measure of the local density of states (LDOS). As the electron beam can penetrate through, e.g., a metallic coating, this approach can be used to measure active material buried inside a nanocavity.
A second approach is to use a pump-probe scheme is which the electron beam is used to probe the material response after excitation with a short laser pump beam. In a semiconductor, the presence of (photo-excited) electrons in the conduction band alters the secondary electron emission yield. Probing the secondary electron yield as a function of spatial position and laser-electron pulse delay allows to map electron migration and recombination at high spatial and temporal resolution. However, in this scheme, carrier trapping, which competes with migration and can play a major role in nanodevices due to the high surface-volume ratio, remains obscured. We have implemented a lock-in detection scheme, where the laser excitation beam is chopped and the secondary electron detection is phase-locked to the chopper frequency. In this way, both ultrafast carrier dynamics and trapping can be visualized in a single experiment.
A second approach is to use a pump-probe scheme is which the electron beam is used to probe the material response after excitation with a short laser pump beam. In a semiconductor, the presence of (photo-excited) electrons in the conduction band alters the secondary electron emission yield. Probing the secondary electron yield as a function of spatial position and laser-electron pulse delay allows to map electron migration and recombination at high spatial and temporal resolution. However, in this scheme, carrier trapping, which competes with migration and can play a major role in nanodevices due to the high surface-volume ratio, remains obscured. We have implemented a lock-in detection scheme, where the laser excitation beam is chopped and the secondary electron detection is phase-locked to the chopper frequency. In this way, both ultrafast carrier dynamics and trapping can be visualized in a single experiment.

Local decay rates measured inside metal-coated nanocavities with time-resolved cathodoluminescence |
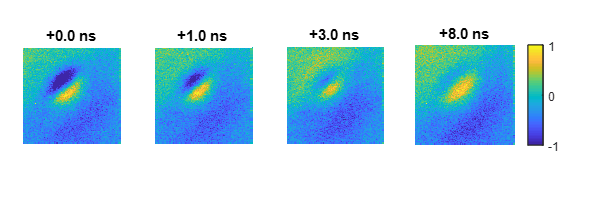
Ultrafast carrier relaxation and surface trapping on GaAs
|
Light-electron-matter interactions
Chemical reactions induced by low energy electrons are important for a wide range of technological and medical applications. In microscopy and lithography, low energy (<20eV) electrons generated upon (electron-beam or EUV) exposure induce a cascade of reactions that lead to structural damage, which crucially define resolution limits. But also in many other applications, for instance radiation therapy, low-energy electrons play a pivotal role. We use fluorescent molecules as reporters for charge migration and reaction dynamics and our integrated microscope allows to study in-situ during irradiation. The presence of and reaction with low energy electrons leads to fluorescence blinking and bleaching, which provides a read-out for the extent and course of reactions. Moreover, initially non-fluorescent organic macromolecules develop fluorescence as a result of electron-induced cross-linking, which also allows to monitor chemical and structural rearrangements during irradiation.
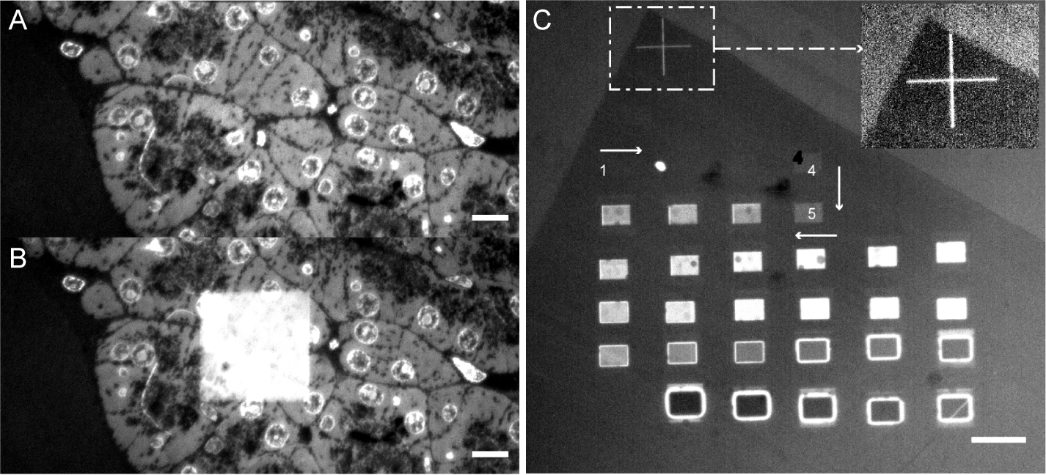
Fluorescence induced by (A,B) electron irradiation of biological material and (C) irradiation with increasing electron fluence of bare polymer
|
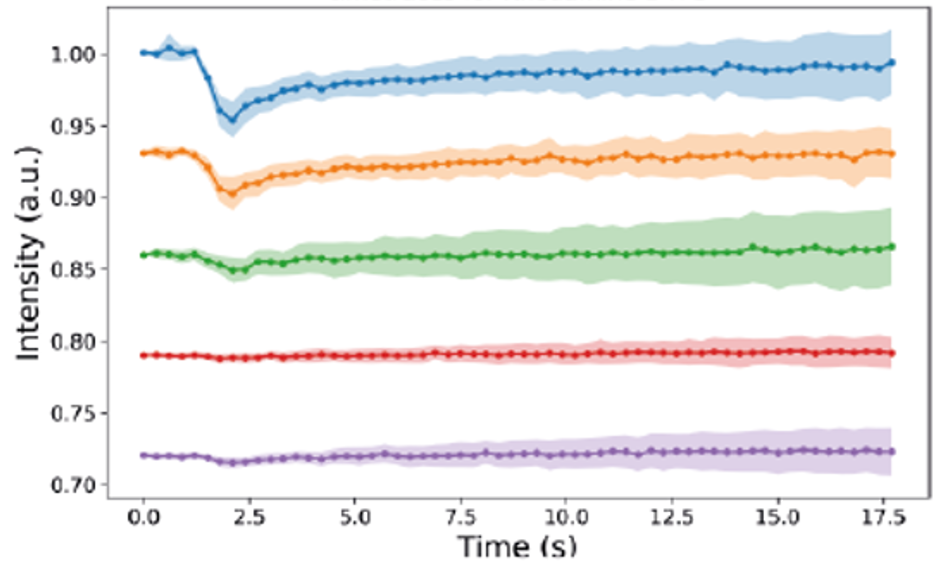
Electron-induced bleaching in a film of fluorescent molecules
|
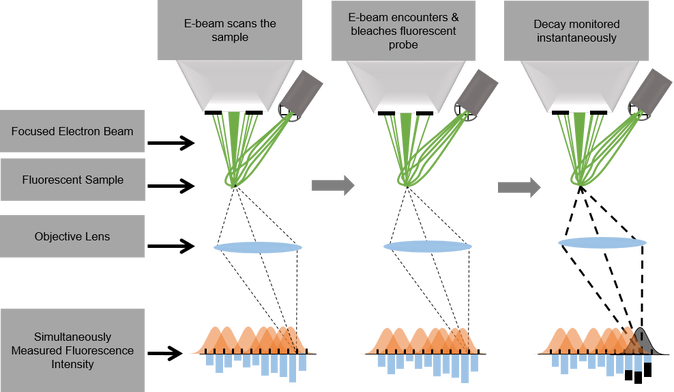
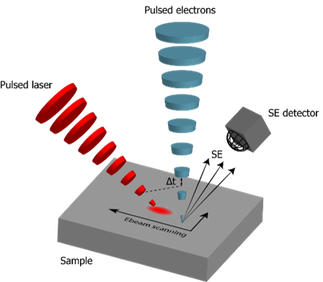
We want to use the electron-light-molecule interaction to better localize molecules in EM images. With CLEM, molecules can be visualized in the electron micrographs, but the resolution gap between both modalities precludes precise localization. Superresolution fluorescence microscopy can be used to bridge the gap, but sample preparation for EM is often at conflict with requirements for superresolution techniques. Moreover, registration errors add to the localization error. In a first approach, we use the fluorescence bleaching induced by the focused electron beam to localize the fluorescence: while the electron beam scans the sample, a decrease in signal is detected on a few camera pixels when the beam bleaches fluorescent molecules. By assigning this decay to the electron beam position, localization is achieved in perfect registry with the simultaneously acquired EM signal (see schematic).